Espace patient
Informations annexes au site
1. Informations générales
La Recherche Clinique est à l’origine de l’ensemble des traitements et médicaments que nous connaissons, que ce soit le paracétamol, les vaccins ou encore les antibiotiques, en passant par les traitements de pointe en cancérologie ou cardiologie. Étape indispensable dans le développement de nouveaux médicaments et traitements, située entre la recherche fondamentale et la mise sur le marché, la Recherche Clinique représente donc un enjeu majeur de santé.
La Recherche Clinique favorise l’accès des patients aux innovations et à la qualité des soins ainsi que la formation continue des médecins investigateurs et de leurs équipes.
Les études
Les études peuvent concerner des nouveaux médicaments ou des dispositifs médicaux (cathéters, etc…), des analyses biologiques ou des nouvelles stratégies thérapeutiques. Celles-ci peuvent reposer sur l’analyse de données recueillies de manière prospective (recueil des données concomitamment à la prise en charge du patient) ou de manière rétrospective (recueil des données à posteriori). Chaque étude est régie par un protocole qui définit ses objectifs, les méthodes pour y parvenir et la population étudiée.
Il existe deux grandes catégories d’études cliniques :
- interventionnelles avec une « intervention » qui modifie la prise en charge habituelle. Cette intervention peut être comparée à la prise en charge habituelle ou à un placebo.
- non-interventionnelles (anciennement observationnelles) sans modification de la prise en charge habituelle.
| EN CE QUI CONCERNE LES ESSAIS MÉDICAMENTEUX, QUATRE PHASES EXISTENT : | |||
|---|---|---|---|
| OBJECTIFS | DURÉE | POPULATION | |
| Phase I | Première administration : étude du devenir dans l’organisme et de la sécurité du médicament | Quelques jours à quelques mois | Petit nombre de volontaires sains ou parfois malades |
| Phase II | Evaluer l’efficacité et la tolérance du médicament. Recherche de la posologie optimale | Quelques mois à 2 ans | Petit groupe de patients atteints de la maladie à traiter |
| Phase III | Phase de confirmation avec évaluation du rapport bénéfice/risque du médicament | Quelques années | Plusieurs centaines à milliers de malades |
| Phase IV | Après la mise sur le marché, évaluation du médicament en conditions réelles d’utilisation et détection d’effets indésirables rares | Quelques années | Plusieurs milliers de malades |
Plusieurs intervenants sont nécessaires pour la mise en place et le suivi d’une étude :
- Le promoteur :
à l’initiative de la recherche, il en assure le financement et la responsabilité de son bon déroulement. Le promoteur peut être institutionnel ou académique (INSERM, société savante, établissement de santé, etc…), ou alors industriel (laboratoire pharmaceutique).
- L’investigateur et son équipe :
L'investigateur peut-être de profession médicale ou paramédicale. Choisi par le promoteur, le médecin va, avec l’aide de son équipe, mettre en application le protocole d’essai clinique. L'investigateur procède au recrutement des patients en leur expliquant le principe de l’essai et son déroulement pour ensuite recueillir leur consentement. L'investigateur et son équipe accompagnent ensuite le patient pendant tout le déroulement de l’étude.
- L’Attaché de Recherche Clinique (ARC) :
il est l’interlocuteur privilégié de chacun des acteurs tout au long du protocole. Il fait le lien entre le promoteur de l’étude et le médecin investigateur. Il veille au bon déroulement de l’étude en s’appuyant notamment sur les normes prévues par le protocole et les bonnes pratiques cliniques.
- Le Technicien d'Études Cliniques (TEC) :
il assiste l’ARC et l’investigateur dans le suivi des patients inclus dans l’étude, il est en charge de la saisie des données et des prélèvements biologiques si besoin.
2. Participation à une étude clinique en pratique
| DIFFÉRENTES ÉTAPES SONT NÉCESSAIRES POUR LA PARTICIPATION À UNE ÉTUDE : |
|---|
| Mon médecin me propose de participer à une étude. Il me présente l’étude de façon claire et loyale : objectifs, bénéfices espérés, risques et contraintes, droit d’accès aux informations. Il me remet une note d’information. |
| L’équipe investigatrice vérifie que je corresponds bien à la population étudiée et qu’il n’y a pas de critères d’exclusion de l’étude. |
| Selon l’étude, mon consentement est recueilli à l’écrit ou à l’oral, et noté dans le dossier médical. |
| Je suis inclus dans l’étude et ma prise en charge suit le protocole de l’étude. Selon les études, une randomisation (tirage au sort) peut être effectuée pour déterminer quel protocole sera appliqué. |
| Je suis suivi au fur et à mesure de ma prise en charge par l’équipe de recherche clinique. La durée de l’étude est variable. |
| A la fin de l’étude, je peux demander à mon médecin les résultats de celle-ci. |
Par ailleurs, plusieurs informations sont importantes en cas de participation à une étude :
- Je ne reçois aucune rémunération pour ma participation à une étude. Il n’y a aucune charge financière. Certaines études prévoient la prise en charge de tout ou partie des frais engagés liés à la recherche (frais de transport par exemple).
- Je peux à tout moment décider d’arrêter l’étude, sans nécessité de justification, et sans que cela ne cause un préjudice pour ma prise en charge.
3. Réglementation en lien avec la recherche clinique et la réutilisation des données de santé
1. Essais cliniques
La Recherche Clinique est réglementée par le code de la santé publique, différents textes de loi, notamment la Loi Informatique et Libertés de 1978 modifiée, la loi Jardé parue en 2012 avec ses ordonnances d’application de 2016, ainsi que par des référentiels de bonnes pratiques cliniques (2006). Toute étude nécessite une autorisation de l’ANSM (Agence Nationale de Sécurité du Médicament et des produits de santé), un avis favorable d’un comité d’éthique ou d’un CPP (Comité de Protection des Personnes) et le respect des recommandations de la CNIL (Commission Nationale de l’Informatique et des Libertés) et du RGPD (Règlement Général pour la Protection des Données) en matière de protection des données.
Les études cliniques reposent toujours sur la participation volontaire des personnes. Pour les études dont le GHBS est promoteur, les activités de recherche s’effectuent sous la responsabilité de l’établissement. L’opposition à cette participation peut être exprimée à n’importe quel moment.
Lors de votre inclusion dans un protocole de recherche clinique mené au GHBS, certaines de vos données personnelles (nom, prénom, date de naissance) pourront être enregistrées dans un logiciel de gestion des essais cliniques hébergé au CHU de Rennes. Ces données sont utilisées à des fins d’organisation de l’activité de l’unité de recherche clinique du GHBS et ne sont accessibles qu’aux personnes autorisées et soumises au secret professionnel.
Si vous souhaitez vous opposer à votre participation à des études ou à l’utilisation de vos données de santé à des fins de recherche, vous pouvez contacter le secrétariat de l’Unité de Recherche Clinique (cf. Contacts).
Celui-ci vous orientera selon votre demande vers les personnes compétentes de l’Unité de Recherche Clinique, ou le délégué à la protection des données (dpd@ghbs.bzh, courrier postal : GHBS, délégué à la protection des données, 5 avenue de Choiseul, 56322 Lorient).
2. Réutilisation des données
En vertu de l’article L. 6113-7 du Code de la santé publique, les données de santé recueillies dans le cadre du soin peuvent être réutilisées pour la production d’indicateurs d’activité pour le pilotage ou la qualité des soins.
Ces données de santé recueillies dans le cadre du soin peuvent aussi être réutilisées, sauf opposition de votre part, à des fins de :
- Recherche,
- Réalisation d’études de faisabilité
Ces données sont issues du dossier médical. Il s’agit de données identifiantes (nom, prénom, âge) et de données de santé (données médicales, biologiques, d’imagerie...).
Ces données sont utilisées afin d’améliorer les connaissances et les pratiques des professionnels de santé au profit des patients.
Cette utilisation est encadrée par le RGPD et s’inscrit dans le cadre légal de la mission d’intérêt public ou de l’intérêt légitime du GHBS selon les situations. Les analyses sont réalisées de façon confidentielle sur des données pseudonymisées, sans mention des noms et prénoms. Les résultats sont produits sous une forme agrégée qui ne permet en aucun cas de vous identifier.
Ces données vous concernant, vous disposez des droits suivants : droit à l’information, droit d’opposition, droit d’accès aux données, droit de rectification, droit à l’effacement ou droit à l’oubli, droit à la portabilité des données, droit à la limitation des traitements. Le délégué à la protection des données (DPD) du GHBS veille au respect de cette réglementation.
Un entrepôt de données de Santé (EDS) est en cours de déploiement. Lorsqu’il sera opérationnel, il rassemblera les données informatisées extraites du SIH (Systèmes d'Information Hospitalier) du GHBS. Ces données entreposées pourront être réutilisées afin de mener à bien des recherches.
Une demande d’autorisation auprès de la CNIL sera effectuée avant de mettre en place cet entrepôt. Le responsable de traitement en sera le GHBS. L’EDS sera administré par l’équipe du Centre de Données Cliniques (CDC) du GHBS. Seules des personnes soumises au secret professionnel pourront accéder à ces données. Leur durée de conservation est définie par l’article R1112-7 du code de la santé publique.
Ces données anonymisées pourront être partagées avec d’autres établissements hospitaliers ainsi qu’avec des partenaires publics ou privés du GHBS pour des collaborations scientifiques, mais aussi être croisées avec les données du Système National des Données de Santé SNDS).
Pour chaque étude utilisant les données de l’entrepôt, l’information des personnes concernées et le respect du RGPD, de la loi informatique et libertés et des préconisations de la CNIL seront nécessaires.
Si vous souhaitez vous opposer à la réutilisation de vos données de santé, vous pouvez contacter le délégué à la protection des données :
(dpd@ghbs.bzh, courrier postal : GHBS, délégué à la protection des données, 5 avenue de Choiseul, 56322 Lorient).
Si après nous avoir contactés, vous estimez que vos droits ne sont pas respectés, vous pouvez introduire une réclamation auprès de la CNIL (Commission Nationale de l’Informatique et des Libertés) : https://services.cnil.fr/.
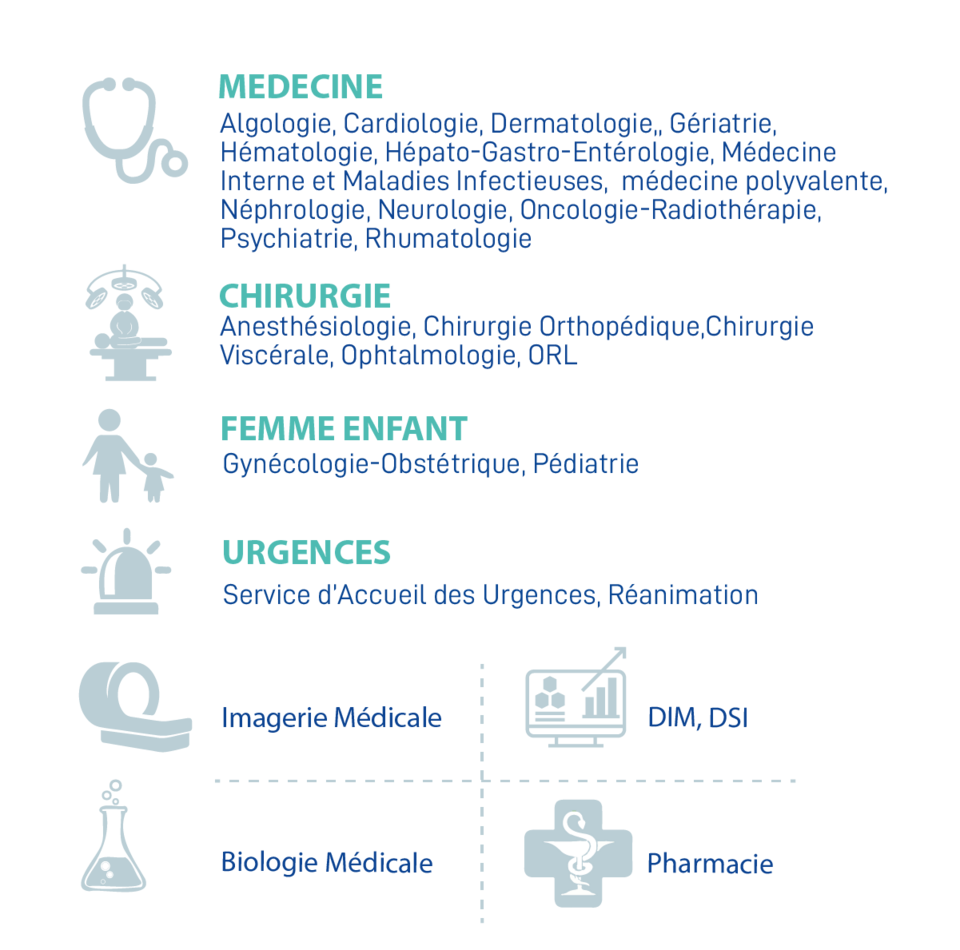
4. Liste des services participant à la Recherche Clinique sur le GHBS
Liste des services participant à la Recherche Clinique sur le GHBS :
5. Etudes réutilisant des données de santé
Actuellement, des études réutilisant des données de santé sont en cours :
- Validation de dispositifs médicaux de diagnostic in vitro (étude Plasmabank, société Diagnostica STAGO SAS)
- Lettre d’information concernant le travail sur la prédiction des parcours des patients non programmés
- « Lettre d’information concernant l’étude ProVAC (CHU de Rennes) sur les pratiques d’alerte des professionnels vis-à-vis des violences à enfant. »
6. Liens utiles
Informations sur la recherche et vos droits :



Registre des essais cliniques :
![]()
Réseau :

7. Interview du Dr Christian SIRE

Le docteur Sire a, dès son arrivée dans l’établissement, oeuvré pour le développement de la Recherche Clinique.
Il rappelle l’importance de cette recherche et la nécessité de l’élargir au plus grand nombre de services, ainsi que l’intérêt pour le GHBS de développer cette activité.
De la même façon, il souligne le bénéfice conséquent, en termes d’image et d’attractivité, pour les établissements qui se consacrent à la Recherche Clinique.
| Quel est, pour un patient, le risque d’être inclus dans un protocole de Recherche Clinique ? |
| CS : Il n’y a pas de risque directement lié à la Recherche Clinique. Le patient bénéficie de traitements qui, s’ils sont encore à l’étude, ne peuvent qu’être bénéfiques. Il a d’ailleurs été démontré que les patients inclus dans les protocoles avaient un meilleur pronostic. Un comité d’éthique valide les différentes étapes d’un protocole depuis la proposition d’ouverture par le promoteur. Pendant toute la durée du protocole, les promoteurs effectuent également des visites dans les établissements. |
| Quelle est la durée moyenne d’un protocole ? |
| CS : La Recherche Clinique requiert de la patience. Par exemple, à mon arrivée en 1994, j’ai commencé une inclusion de patients dans un protocole. Les derniers patients ont été inclus en 1999 et le fruit des travaux publiés en 2003. Il faut compter généralement une dizaine d’années pour une recherche d’importance. |
| À ce sujet, comment réagissent les patients quand on leur propose d’intégrer un protocole de Recherche Clinique ? |
| CS : En 15 années de Recherche Clinique, à raison de 30 à 40 patients inclus par an, je n’ai eu que quelques rares refus. Il est bien évident que le patient acceptera facilement son inclusion si on prend le temps et le soin de lui expliquer clairement et précisément la nature du protocole et les avantages qu’il peut en tirer. |
| Quelle est l’importance de la Recherche Clinique pour le GHBS ? |
| CS : La Recherche Clinique est extrêmement importante pour un établissement comme le nôtre. Elle lui offre, au travers des publications de nos travaux dans des revues à portée internationale, une exposition sans pareil. Elle permet également de nous donner la connaissance et l’accès aux toutes dernières avancées ; ce qui est un plus indéniable pour le patient. Le fait de faire de la Recherche Clinique permet également de recruter plus facilement des jeunes praticiens. |
| Que souhaiter pour améliorer encore la Recherche Clinique au GHBS ? |
| CS : Au vu de la durée d’un protocole et du travail énorme qu’il représente, il me semble important d’avoir une personne qui assure, en plus des ARC, la coordination en interne dans l’établissement et avec le promoteur. Cette personne aurait également une mission de communication tant en interne que vers l’extérieur. Il est également indispensable que l’établissement communique sur son activité de Recherche Clinique. |